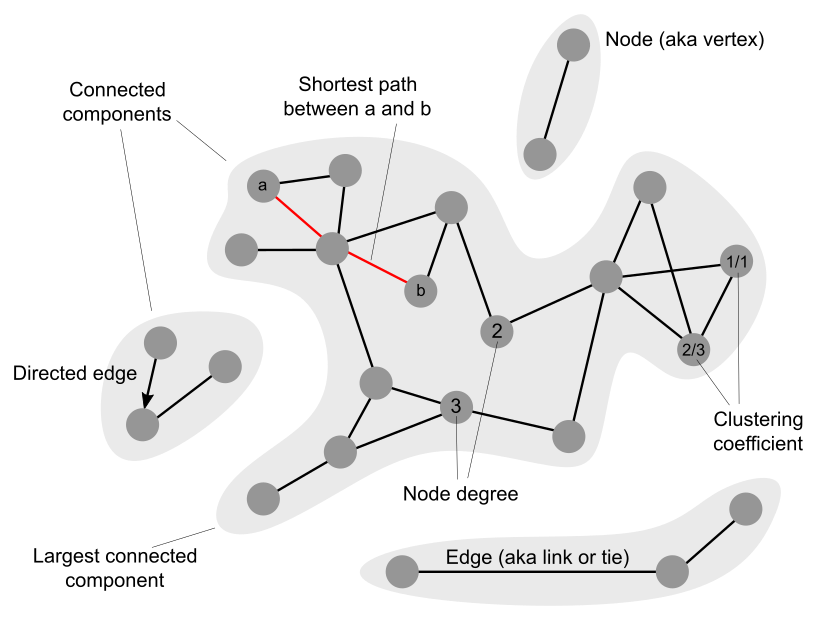
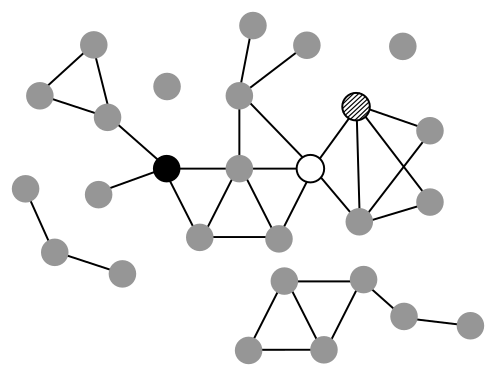
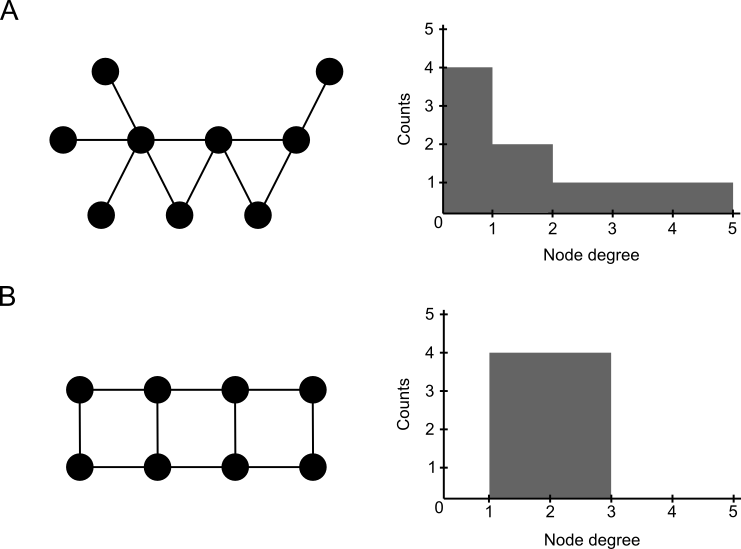Exercise
Consider the following hypothetical scenario:
The University Medical Centre at JGU-Mainz is carrying out a study on children with Progeria, an abnormal congenital condition characterised by premature aging.
Progeria's most evident manifestations are premature greying, hair and hearing loss, cataracts, arthritis, wrinkles and loose skin. The latter being the result of
muscle and skin cell senescence.
Based on different screenings involving sick and healthy children, researchers at UniMedizin have identified 44 genes associated with Progeria and have asked you
to analyse them in order to better understand the molecular basis of this disease and pinpoint potential drug targets.
Armed with the Network Biology tools that you've just learnt, you are set to help them out.
1. Use HIPPIE's Network Query to visualise high-confidence interactions
within the set of Progeria genes from UniMedizin. Verify that there is indeed a significant association between these genes and Progeria.
2. Identify the top-3 hubs in the Progeria subnetwork. Study their function in UniProt. Are these proteins functionally related?
What could their role be in Progeria?
3. Identify the node with the highest clustering coefficient in the Progeria subnetwork. Study its function and the function of its direct partners in
UniProt. Can you deduce the function of this protein complex? (hint: you don't really have to calculate clusterings
here, instead look for closed-triangle motifs in the network)
4. The UniMedizin researchers tell you that they're particularly interested in the Zyxin protein (ZYX), because its function is not well-known. Can you infer it by analysing the function
of its partners in the Progeria subnetwork?
5. What's the size of the largest connected component (LCC) of the Progeria subnetwork? Taking random sets of 44 proteins from the human protein interactome
results in LCCs with an average size of 3. What does this difference tell you about the inter-connectedness of the Progeria subnetwork?
6. The average shortest path length of the human protein interactome is approximately 5. The average shortest path length of the Progeria subnetwork is slightly larger than 3. What can you
deduce about the importance of this difference?
7. Previous research has pointed at protein LMNA as one of the most important factors behind Progeria. This protein is crucial in nuclear assembly, nuclear membrane formation and telomere dynamics.
Study the function of the direct partners of LMNA in UniProt. Do these interactions support the importance of LMNA in this disease? Why?
8. The role of LMNA in telomere dynamics intrigues you. You know that at every cellular division telomeres get shorter, up to the point where they can't shrink anymore and cells enter a state called
cellular senescence. You suspect that this might be connected to the molecular basis of Progeria. Are there any differences in telomere length between Progeria patients and age-matched children?
(hint: do a Google search for progeria and telomere length).
9. You now have three important pieces of information for your final report for UniMedizin: the function of topologically important nodes in the Progeria subnetwork, your analyses about the
inter-connectedness of the subnetwork and the telomere length involvement in aging. Can you put these three pieces together and speculate about the biological processes that are affected by mutations
in the 44 genes identified by your colleagues at UniMedizin?
10. What kind of molecular functions could be the target of a new therapy against Progeria, based on your network analysis?