| Input
- What is ProteinPathTracker?
ProteinPathTracker is an easy-to-use web tool that allows to track the evolutionary history of a query protein.
-
- For what should it be used?
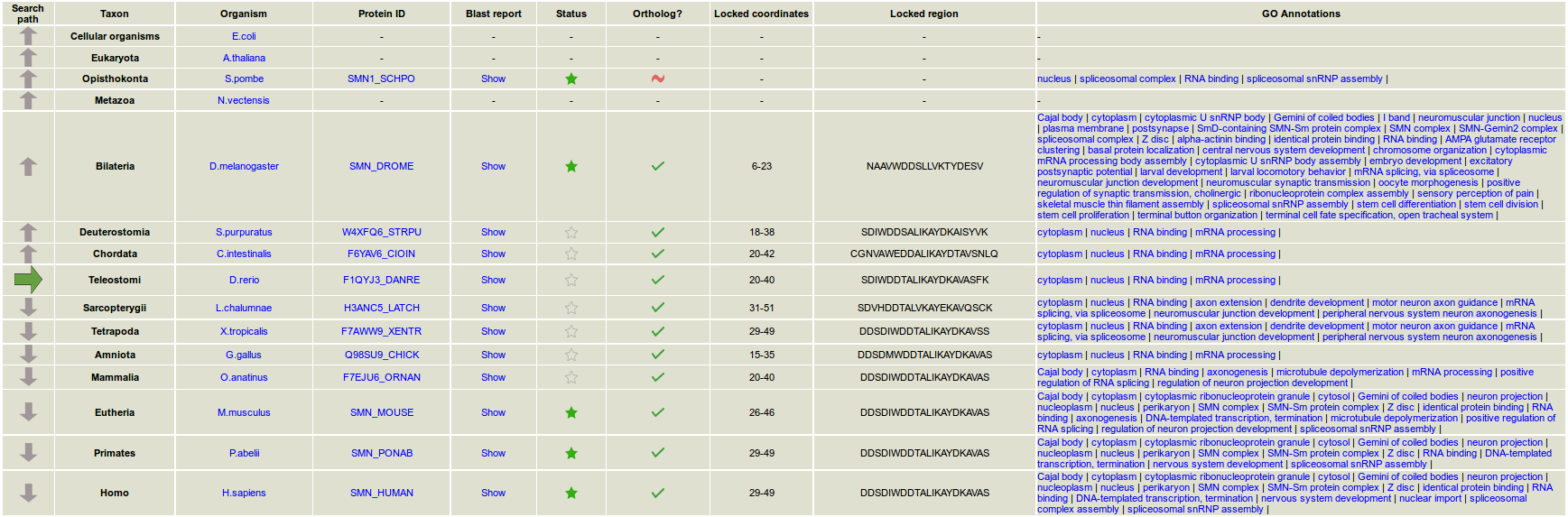
To study the path through evolution of a query protein. ProteinPathTracker tracks the homologs of a query protein in one of the six available paths, and provides their GO terms. The study of the homolog's annotations may serve to know how did the protein evolve in the selected proteomes.
-
- For which type of user is it prepared?
ProteinPathTracker is designed to look as simple as it can get, so that any user may execute it. More advanced functionalities like locking a region are optional.
-
- Which datasets are used?
193 complete reference proteomes were downloaded from the UniProt Proteomes section on 27th April 2017. These proteomes belong to different taxonomic groups. GO terms for each protein were obtained from their individual UniProt entries. The proteomes are distributed in six evolutionary paths:
- cellular organisms → Homo
- Primates → Homo
- Viridiplantae → Arabidopsis
- Fungi → Schizosaccharomyces
- Bacteria → Escherichia
- Arthropoda → Drosophila
-
- How should you use ProteinPathTracker?
ProteinPathTracker needs just a protein sequence in FASTA format to execute, either pasted or uploaded. The rest of the parameters are optional. After providing the protein sequence, click in "Track ProteinPathTracker, track!" to start its execution. Results will appear in the same window in less than one minute.
-
- What does the "evolutionary path" mean?
As the web tool looks for homologs through evolution, the evolutionary path shows the proteomes in which homologs are searched. By default, the selected proteomes are used, but in some taxa the user may change the desired proteome.
-
- Which parameters can be modified to start the execution of ProteinPathTracker?
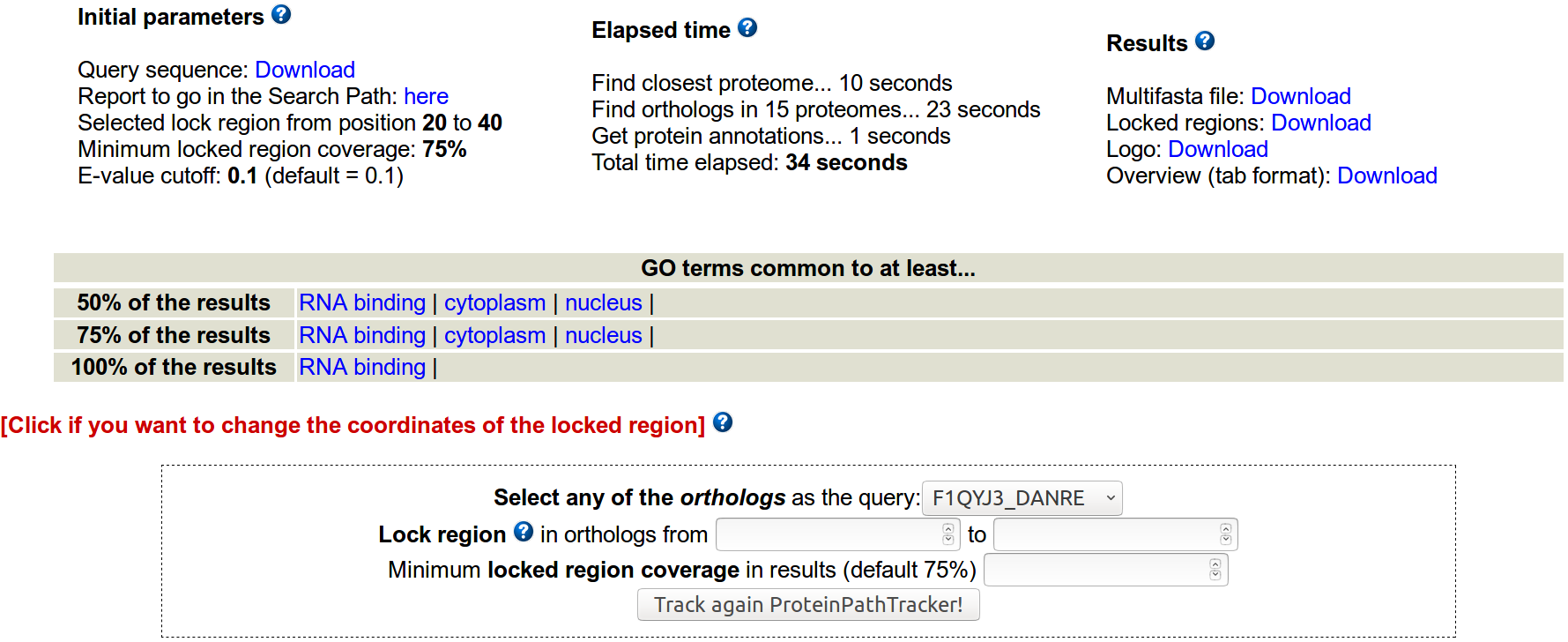
Evalue cutoff to study the significance of the Blast searches can also be modified (default evalue = 0.1).
By default, the lock region strategy is off, but it can be turned on by selecting the coordinates of the lock region to follow in the homologs. The minimum length of any locked region is 10 aminoacids. If the user locks a region, the minimum %coverage of the selected locked region in the results can also be specified. By default, a minimum of a 75% coverage of the initial locked region must be met to consider positive a region.
-
- What does the functionality "lock region" mean?
The evolution of a particular subregion is followed throughout the protein path in evolution, via its mapping in the different homologs.
-
- When should you lock a region?
When you are interested in a specific motif or domain in the query protein. It allows the tracing of a domain in, for example, multidomain proteins. Locking an annotated region with a particular function may help in assessing whether the homologs share it or not, and even when it appeared in evolution.
-
- Why did you not get any locked regions?
If you didn't get any locked region, even if you selected it, try by selecting a different region or query (amongst the orthologs found), or by modifying the minimum locked region coverage (75% by default).
| |